

NOMIS Research Projects
NOMIS research grants support unconventional research projects led by researchers who have demonstrated exceptional scientific capabilities and leadership. The grants enable scientists to spearhead pioneering research to answer bold questions and advance yet unexplored approaches across scientific and academic disciplines. We award research grants to investigators with an excellent track record in leading groundbreaking, high-risk basic research.

Details
The Question
Plants thrive in constantly changing environments, but unlike animals, they cannot move to find food or escape unfavorable conditions. Instead, they rely on highly efficient cellular stress responses and, most importantly, the ability to adjust their growth to reach favorable areas while avoiding unfavorable ones. This precise directional growth is particularly pronounced in roots, which explore the soil to locate water and nutrients, optimizing their uptake from the most favorable soil patches. These capacities depend on the ability to perceive the local environment and make growth decisions. However, beyond responding to basic cues like light and gravity (which have been documented and explored), can plants sense their environment in a way that resembles how animals use smell and taste — abilities that help animals locate resources and avoid dangers?
Surprisingly, this question can’t yet be answered as very little is known about how plant roots detect and respond to the vast array of chemical cues in the soil that carry information about the local environment. However, plant genomes encode hundreds to thousands of receptor kinases — a class of proteins known for recognizing chemical signals and triggering cellular responses. Yet, the function of nearly all these receptors remains a mystery. The Mapping the Root Perceptome project hypothesizes that, much like animals, plants use these receptors to detect a wide range of yet unidentified chemical cues, guiding their growth decisions in response to their environment.
“My research could transform how we think about plant life, opening new paths for sustainable agriculture, improving ecosystem resilience, and advancing climate change solutions. By comprehensively decoding how plants sense their environment, we will better understand their enormous success on our planet and can better harness their potential to support life on Earth.”
— Wolfgang Busch
The Approach
The Mapping the Root Perceptome project aims to map the chemical world that roots can perceive — that is, their “perceptome” — and investigate whether roots function as sensory organs with specialized structures for detecting and interpreting chemical information, similar to how tongues and noses perceive and process signals in higher animals. By systematically testing how thousands of chemicals influence root growth and identifying the root structures and receptors involved in perception, the researchers hope to uncover the molecular and cellular mechanisms by which roots sense and respond to external cues.
Discoveries resulting from this work will significantly advance our understanding of how neuronless organisms like plants perceive and interact with their surroundings. This knowledge could have practical applications in agriculture and environmental sustainability, helping to develop plants with enhanced resilience to changing conditions, improved nutrient uptake, increased carbon storage in the soil, and even engineered root systems tailored for specific environments. Just as breakthroughs in human and animal sensory biology have revolutionized medicine and technology, this research will reshape how we think about plant life — revealing the hidden sensory sophistication of roots and their capacity to navigate and adapt to their environment.
The Mapping the Root Perceptome project is being led by Wolfgang Busch at the Salk Institute for Biological Studies in La Jolla, US.
Feature image: Laser confocal microscopy image (maximum intensity projection) of an Arabidopsis thaliana root tip. Cell walls are visualized in yellow/purple and nuclei appear as bright yellow spots. (Photo: Salk Institute)

Details
The Question
Clouds play a critical role in climate change. By reflecting sunlight back into space, they cool the planet, while simultaneously trapping infrared radiation in the atmosphere, contributing to warming. These opposing effects shift as the climate warms, making clouds a key factor in determining how much the planet will ultimately respond to increasing greenhouse gas concentrations.
Yet, despite decades of intensive research, quantifying how clouds respond to global warming remains a challenge. This lack of understanding of clouds is the greatest unknown in climate projections. The difficulty of representing clouds in atmospheric models leads to significant uncertainties in weather forecasts. In particular, accurately forecasting extreme precipitation events associated with deep convective clouds remains a major hurdle.
At the heart of this challenge is the complexity of cloud formation, which depends on both microscopic processes, like droplet and ice crystal formation, and large-scale atmospheric processes, such as low- and high-pressure systems spanning hundreds to thousands of kilometers. Accurately representing the physical laws that govern all these processes is currently infeasible, even for the most advanced supercomputers.
“With Deepcloud, we are breaking one of climate science’s biggest barriers: understanding and representing cloud processes in a warming world. By uniting advanced AI, open data and community-driven research, we’re transforming how clouds are modeled — and with them, the future of climate prediction.”
— Markus Rex
The Approach
The Deepcloud project aims to overcome this challenge by using machine learning to model cloud processes. Recent progress in artificial intelligence approaches, combined with the recent quantum leap in the availability of detailed cloud data, open up new possibilities for understanding and simulating clouds.
Rather than explicitly describing all physical processes within clouds, the machine-learning model will learn the most efficient representation of how small-scale processes influence cloud behavior. To train the model, the project will leverage unique observational data from two key sources: a recent year-long Arctic expedition and a cloud-observing satellite launched in 2024. This model of cloud processes will be used to explore how Arctic clouds respond to global warming and how increases in small particles in the atmosphere influence cloud properties. The researchers hope to uncover the extent to which cloud ice is replaced by cloud liquid in a warmer climate and how changes in cloud condensation nuclei and ice nucleating particles affect cloud processes and cloud properties. These insights will advance our understanding of cloud-related climate feedback at high latitudes, transforming the future of climate prediction and policy, ultimately safeguarding the planet for future generations.
Deepcloud: Tearing Down a Long-Standing Barrier in Climate Research by Pioneering a New AI-Based Model Approach is being led by Markus Rex at the Alfred Wegener Institute, Helmholtz Center for Polar and Marine Research (AWI) in Potsdam, Germany.
Feature image: Convective clouds in the clean air of the tropical West Pacific. (Photo: AWI)

Details
The Question
Fueled by powerful new telescopes and the discovery of thousands of exoplanets — planets that orbit stars other than the sun — the search for life beyond Earth is accelerating. Yet whether in the solar system or on distant exoplanets, finding life may require a broadened perspective. Could life exist in Venus’ acidic clouds, on airless planets or in other extreme environments once considered uninhabitable?
Many known exoplanets are too warm for surface liquid water and therefore considered inhospitable to life. Liquid is a fundamental requirement for life as we understand it, but whether that liquid must be water is not known.
Groundbreaking experiments in Sara Seager’s lab have shown that some key building blocks of life remain stable in sulfuric acid — the main component of Venus’ clouds — challenging conventional thinking. Beyond Venus, the researchers suggest that ionic liquids — special nonevaporating liquids that can be hospitable to biomolecules — can form and exist naturally, perhaps even on planets without atmospheres.
“My research expands the definition of habitability, offering new pathways to detect life on planets once dismissed as inhospitable. At a deeper level, it speaks to a universal human desire to understand our place in the cosmos — and the aim to discover that we are not alone. While driven by pure scientific curiosity, this work can occasionally lead to practical spinoffs, especially through the development of hardware for space missions, as my team is actively doing.”
— Sara Seager
The Approach
The project From Lab to Cosmos aims to expand the definition of habitability by conducting laboratory experiments that study biomolecules’ chemistry in sulfuric acid for Venus missions; explore biomolecules in ionic liquids as exotic solvents; and predict signs of life based on planets that could sustain life in extreme environments.
The most transformative potential of this research lies in its implications for the origin of life. By examining exotic solvents and rejecting water as the sole prerequisite for biology, we gain access to a wider chemical space — one that may encompass the primitive pathways by which life began. These pathways, now erased from Earth’s surface record, may still be active elsewhere, and exploring them could redefine our understanding of life’s beginnings and its potential distribution across the cosmos.
From Lab to Cosmos: Rethinking Habitability and the Search for Life Beyond Earth is being led by Sara Seager at the Massachusetts Institute of Technology (MIT) in Cambridge, US.
Feature image: Left photo courtesy of Sara Seager; center photo by JAXA; right photo by NASA
NOMIS Researcher(s)
NOMIS Project 2025
— 2028

Details
The Question
Long before the advent of ChatGPT and other large language models (LLMs), the question arose: Can a black box think? The research project Assisted Thinking: A Deep History of Scholarly AI meets Alan Turing’s (1950) seminal challenge with the assumption that intelligence is distributed between humans and machines. It pursues two lines of inquiry: on the one hand, by writing the history of AI as a genealogy of thinking machines from the baroque to the present, and on the other hand, by using insights gained from this genealogy to critically examine the use of language in current LLMs. The project’s guiding questions probe assistance and creativity in intellectual work by examining the architecture, modes of interaction, and language use of both historical and current thinking machines with a genealogy from writing desks and scholarly devices since the baroque period to current LLMs. How does the architecture of those intellectual furnishings facilitate thought? How is language incorporated in the device? Finally, how can the usages of the (historical) machines be assessed while they produce both creativity and constraints?
The Assisted Thinking project also will explore other questions, including, what does this prehistory of LLMs look like? What are the key scenarios in this long trajectory of scholars’ interactions with their thinking aids? Who is involved in the deeper history of artificial intelligence, reaching back far beyond the famous Dartmouth conference in 1956, when the term “artificial intelligence” was coined?
The Approach

Following critical AI studies with a media historical perspective, the core concept of “assistance” takes the agential relationship between human and AI to be an unequal but distributed phenomenon, circumventing the discourse of the machinic replacement of human beings. The assisting device acts as a moderator and facilitator of human ideas and knowledge production, and it is aimed at creativity, understood here as the productive outcome of a shared interaction rather than as the characteristic of a single participant.
Two foci elucidate these concepts from different historical and conceptual angles. While the “Deep History of AI” investigates the assistive function of “intellectual furnishings” like desks and writing cabinets from the baroque to the present, the focus on prompts as the interface between human and machine examines the change of language use while becoming both a medium of assistance and an agent in the creative interaction with the technology.
The Assisted Thinking project is being led by Markus Krajewski at the University of Basel (Switzerland).
NOMIS Researcher(s)
NOMIS Project 2025
— 2027

Details
The Question
Imagine you receive this message from a friend: “It finally happened, I got the confirmation letter in the mail this morning, let’s go out tonight to debrief.” As soon as you read this, your brain starts building a complex model: You might immediately know that your friend is talking about a job offer they have been waiting for, for over a year, and that they will be both excited and nervous, because the job means moving countries and their partner doesn’t want that; you might immediately think about whether you can move your 6pm yoga class tonight to go and meet your friend, or whether it might make sense for you to pick them up from home on your way back from work, or whether they are working in their office today.
When we talk with our friends, listen to the news or make plans with colleagues, our brains make sense of these complex situations by using background knowledge to create rich and powerful inferences about the mental states of other people. We are constantly reasoning about the things they know, their likely goals, whether to trust another person, whether to change our minds and when to make new plans together or seek new information.
This kind of social reasoning is central to human life and vital to the buildup of knowledge and cohesion in social discourse. Without reasoning about other minds, our ability to learn and share information would be critically limited, yet these core cognitive processes go beyond what contemporary models in modern cognitive science and neuroscience are capable of capturing. Even the most powerful modern artificial intelligence systems lack many aspects of social reasoning, and we currently lack models and methods that would help us to address this fundamental limitation.
The Approach
The project How Reasoning About Other Minds Supports Human Culture: Large-Scale Experimental Simulations of Cultural Evolution With Human Participants aims to fill this gap by conducting large-scale behavioral experiments in which people engage in social interactions with strangers to solve problems, make plans and discover new concepts together. These experiments will leverage new methods in experimental psychology to create situations that expose the rich structure of human social reasoning, and leverage computational methods to extract structure from large-scale behavioral data. Using modern experimentation, the project’s researchers will be able to construct networks of interacting participants who learn from each other over time, and study the evolution of participant behavior in micro-societies.
The experiments will generate large-scale datasets that can be used to test the predictions of formal computational models of social reasoning, and to improve machine learning systems that currently lack many forms of social reasoning. These insights will not only help us understand one of humanity’s key cognitive skills, but will also help to inform the design of digital systems that better support constructive discourse and learning in modern digital discourse arenas, such as social media platforms and conversational AI systems.
The How Reasoning About Other Minds Supports Human Culture project is being led by Bill Thompson at the University of California, Berkeley (USA).
NOMIS Researcher(s)
NOMIS Project 2024
— 2029
Details
The Question
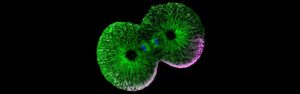
The transition from single-celled to multicellular organisms marks a pivotal moment in the evolution of life on Earth. It has been suggested that this transition may have begun when single-celled organisms failed to undergo complete division, resulting in daughter cells that remained interconnected through their cytoplasm. Notably, multicellular organisms initiate their development with a fertilized egg that undergoes a series of rapid cell divisions, which in some species are incomplete. This process bears a striking resemblance to the proposed mechanisms that led to the emergence of multicellularity. However, it remains unclear how these incomplete divisions can produce cells with distinct forms and functions — an essential characteristic of multicellular organisms.
The Approach
To address this question, the project Cytoplasmic Self-Organization in Early Animal Development will investigate how cells acquire distinct forms and functions in embryos undergoing either complete divisions (ascidians — the closest invertebrate relatives of vertebrates) or incomplete divisions (zebrafish — a popular vertebrate model organism) at the onset of their development.
The researchers’ initial observations suggest an intriguing possibility: In embryos with incomplete divisions, the cytoplasm can self-organize into cell-like compartments that exhibit characteristics typical of intact cells. This implies that the formation and differentiation of cells in multicellular organisms may be driven by this self-organizing property of the cytoplasm rather than solely by the mechanisms of cell division. This hypothesis will be tested through experiments that modify the extent of cell division in both zebrafish and ascidian embryos, exploring whether and how cytoplasmic self-organization can compensate for the lack of division in these scenarios.
The project is being led by Carl-Philipp Heisenberg at the Institute of Science and Technology Austria (ISTA).
Feature image: Nuclei visible via DAPI staining (Photo: Heisenberg lab).

Details
The Question
How do we see in 3D? How our brain does this remains a mystery. All the brain receives are two 2D images on the retina, and yet it somehow constructs a single unified 3D percept. While we know how we would design a visual system with these inputs, the reality is that human 3D vision doesn’t behave as we would expect.
Solving this question has a number of significant implications. First, it helps us to understand the human brain. 3D vision is one of the classic paradigms through which general principles about the brain are formulated and tested, from single-cell recordings in the 1960s, to computational approaches in the 1980s, to probabilistic models in the 1990s. Second, public health. Roughly a billion people worldwide (one-eighth of the world’s population) have 3D vision deficits, so understanding the mechanisms underpinning 3D vision is key. Third, artificial intelligence. We are able to act and navigate in the 3D world in a way that far surpasses existing AI with a fraction of the computational power. How does the human brain do it? Fourth, virtual and augmented reality. We can only build a convincing virtual replica of reality if we know what is required to convince human vision in the first place. Fifth, art, architecture, and spatial design. They all appeal to principles of how we perceive the 3D world.
The Approach
The New Theory of Visual Experience project seeks to unravel the seemingly simple question of how we see in 3D. To understand any system, it is helpful to understand what inputs it responds to. The 3D Vision researchers will thus begin by testing how different visual inputs lead to different 3D visual experiences through behavioral experiments. For example, 3D vision isn’t informed by eye rotation, a mechanism that was thought to be important for linking the retinal image to the world. More recently, the project’s principal investigator developed the “Linton Stereo Illusion” to show that 3D vision directly responds to the projections on the retina rather than reconstructing the position of points in the world.
Once these distortions of visual space have been appropriately quantified, the researchers will build computational models of human 3D vision, collaborating with Nikolaus Kriegeskorte at Columbia’s Zuckerman Mind Brain Behavior Institute.
Finally, they will look for evidence of these processes at work in specific regions of the brain. Bringing together 14 researchers from the US and Europe, the New Theory PI will lead a Generative Adversarial Collaboration titled “Is V1 a Cognitive Map?”
The New Theory of Visual Experience project is being led by Paul Linton at the Italian Academy for Advanced Studies, Columbia University (New York, US).
Feature image: Ann Veronica Janssens: yellowbluepink exhibition, Thomas SG Farnetti. (Source: Wellcome Collection [CC BY-NC 4.0])
Details
The Question
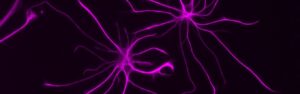
The nervous system and the immune system have historically been studied in siloes. Crosstalk between the two systems — and the role this intersection plays in health and disease — has long been understudied. However, the interplay between the nervous system and the immune system is critical. For example, increasing evidence indicates that inflammation is associated with neurodegeneration in aging, and that a person’s brain function and mental state can affect immunity. Disorders such as Alzheimer’s disease (AD), multiple sclerosis (MS), long COVID and some forms of cancer are thought to result from a combination of damage to the nervous system and resulting inflammation. Yet the cells, molecules and mechanisms that guide these connections are mostly unknown. To better prevent and treat diseases, we need to better understand at all levels how they develop and progress.
The Approach
Through its Neuroimmunology Initiative within the NOMIS Center for Immunobiology and Microbial Pathogenesis, Salk scientists are working to develop a deep understanding of the crosstalk between the immune and nervous systems and the role it plays in health and disease. The Neuroimmunology Initiative encompasses three interconnected research programs:
Body to Brain: Impact of peripheral inflammation on brain cell function
In this line of research, the team will determine how different types of inflammatory events originating in the periphery — such as viral infection, aging and cancer — impact different brain cell types and regions. Additionally, they will investigate the extent to which immune cells, such as T cells, infiltrate the brain and the spinal cord following infections caused by various pathogens. This research program will provide, for the first time, (1) a spatial map of how the nervous system reacts to particular types of inflammation and cytokines, (2) the brain regions in which immune cells infiltrate and establish long-term residency after infection, and (3) the extent to which recurrent infections induce long-lasting changes in the brain’s cellular composition and foster “inflammatory memory” through the induction of epigenetic alterations in neurons and glial cells.
Within the Brain: Impact of immune signaling on brain resident cells
This line of investigation will unravel how resident brain cells, namely microglia, astrocytes and neurons, interact and influence each other during inflammatory events. The researchers will assess how this cellular crosstalk impacts brain inflammation, function and cognitive performance. They will also investigate how the immune cells that infiltrate the nervous system in response to peripheral infection interact with and impact resident brain cells and alter the inflammatory milieu and cerebrospinal fluid in the brain. Finally, they will selectively deplete specific immune cell subsets, such as memory T cells, to delineate their contribution to sustaining brain inflammation over time. Through this multifaceted approach, the team aims to provide insights into the intricate dynamics between infection, immune response and brain homeostasis. Moreover, it may unveil therapeutic targets to mitigate chronic brain inflammation, which is associated with aging and neurodegenerative diseases such as AD.
Brain to Body: Impact of neural signaling on peripheral immunity
This research will look at the flip side of the coin: how the nervous system (brain and spinal cord) senses and influences inflammation and immune responses elsewhere in the body. The scientists will first use neural tracing techniques to identify specific cell types in the brain and spinal cord that respond to inflammatory events such as infection or cancer, illuminating the neurons that relay the health status of an organ or tissue to the nervous system. They will then use genetic and pharmacological manipulations to investigate the roles of those neurons in modulating the immune responses. By activating different stress pathways (e.g., such as those regulating depression and chronic stress) in the brain and spinal cord they hope to determine whether an individual’s mental state impacts immune responses. The researchers will construct a road map of neuronal circuits and interoceptive signals that distinguish between different types of immune responses occurring in different organs. In addition, they seek to deduce how different types of neuronal stress responses impact the initiation and resolution of immune responses in the periphery.
The Neuroimmunology Initiative gives scientists the freedom to tackle currently unaddressed scientific questions, which will open fundamentally new areas of scientific inquiry across human health and disease. Ultimately, this work paves the way toward innovative therapeutic interventions for a wide range of disorders that have both a neurological and immunological component.
The project is being led by Susan Kaech and Nicola Allen at the Salk Institute’s NOMIS Center for Immunobiology and Microbial Pathogenesis in La Jolla, US.
Feature image: Astrocytes (Photo: Salk Institute)
Details
The Question
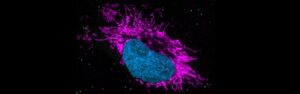
A first line of defense against pathogens, innate immunity is essential for life, providing an immediate but nonspecific response through physical barriers, immune cells and various proteins. It recognizes and responds to a broad range of microbial threats using pattern recognition receptors to detect conserved pathogen-associated molecular patterns.
This complex response demands precision. The activity of innate immune signaling must be properly regulated to orchestrate a transient and balanced response. Errors in controlling innate immunity can lead to disease: Inflammation caused by the aberrant activity of innate immune receptors is now being viewed as a major driver of several human diseases as well as natural aging. But the exact functioning of the innate immune response is still not fully understood.
“My inspiration to pursue basic research stems from a combination of curiosity, a passion for discovery, and the desire to contribute to the foundational understanding of biological phenomena.”
—Andrea Ablasser
The Approach
The Exploring Innate Immune (In)activities project aims to systematically study molecular checkpoints that control innate immune function. Using an interdisciplinary approach, the researchers seek to identify allosteric sequence motifs within immune receptors and signaling proteins that limit overall activity and control the strength, duration and resolution of immune responses. They will use a combination of structural biology, in silico protein prediction algorithms, biochemistry, and cellular assays to establish a mechanistic understanding of allosteric control at a given site and will classify via in-depth phenotypic analyses in cells and in vivo their impact on cell state and tissue inflammation. The project seeks to generate insights into the rules that govern the functioning of innate immunity and to establish the landscape of the regulatory, evolutionary and therapeutic potential of immune signaling pathways.
Exploring Innate Immune (In)activities is being led by Andrea Ablasser at the Swiss Federal Institute of Technology in Lausanne (EPFL; Switzerland).
Feature image: Airyscan imaging of innate immune stimulated HeLa cells with the nucleus marked in blue.
NOMIS Researcher(s)
NOMIS Project 2024

Details
The Question
Messenger RNAs (mRNA) were discovered in the mid-20th century as molecules that copy the genetic information from DNA in the cell nucleus and carry it to the cytoplasm for protein synthesis. Even today, mRNAs are depicted as simple, short linear chains of nucleotides. Yet, this is misleading, as it does not convey the considerable physical length of these nucleic acid polymers, their propensity to fold, and their existence within diverse and variable protein coatings.
Over the course of its life cycle, every mRNA within eukaryotic cells assembles a complex and dynamic protein coating. These supramolecular assemblies, called messenger ribonucleoproteins (mRNPs), protect the integrity of the mRNA, aid in transport and control the spatiotemporal translation into protein. This is especially important in neurons, in which the unique morphology requires that specific mRNAs travel to the most extreme ends of the cells, the axon terminals, for local translation at the synapses. This spatial choreography supplies the synaptic structures with the proteins needed to sustain the signal that directs activities such as proper brain development or storing memories. Despite their importance in gene expression processes, and in contrast to other classes of RNPs such as ribosomes and spliceosomes, the makeup and transformations of individual mRNPs remain a mystery.
“Understanding the architectural principles of mRNA particles will inform us how the fragile genetic information carrier, mRNA, can safely traverse the crowded and extensive cellular environment of neuronal cells, ensuring that precise protein synthesis occurs only upon reaching the correct location.”
—Elena Conti
The Approach
The research project Visualizing the Messenger: Deciphering the Architecture of Neuronal mRNA Particles at the Atomic Level aims to discern the structure and biochemical nature of mRNPs in the brain throughout the mRNA life cycle. Recent breakthroughs in ad hoc biochemical approaches pioneered by the Conti research group have minimized issues with mRNP fragility and variability, allowing for the isolation of endogenous intact mRNPs.
The project will employ genetic editing to enable strategic molecular cloning in a scalable platform with human induced pluripotent stem cells to extract and analyze large quantities of physiological assemblies in their entirety. The researchers will characterize the protein and mRNA content of selected mRNP targets and study their intra- and intermolecular interactions using mass spectrometry and RNA sequencing. The structural organization of the mRNPs will be determined through cryogenic electron microscopy and electron tomography, aided by computational approaches. Elucidating the structure of these mRNPs will provide insights into the mechanisms with which they function in neuronal processes and will facilitate a molecular understanding of how mutations in mRNP components lead to neurological diseases.
The Visualizing the Messenger project is being led by Elena Conti at the Max Planck Institute of Biochemistry (MPIB) in Munich, Germany.
Feature image: Structural basis for the assembly and recognition of modules in mRNA ribonucleoproteins (mRNPs).
NOMIS Researcher(s)
NOMIS Project 2024

Details
The Question
How do human beings’ experience of pictures, such as individual artworks or cultural styles of pictorial representation, shape the ways in which people see the natural and social world beyond pictures? Understanding this experience has the potential to impact fields ranging from education and clinical psychology to social behavior and ethics, marking a significant shift in how we understand the relationship between pictorial art and visual perception.
It has been argued that things in the world — objects and states of affair — come to resemble pictures of them that people have already seen in the past. Objects, then, become partly “depictured”: A real landscape might become Cézannesque when shaped by one’s viewing of pictures of Provence by Paul Cézanne; a real cityscape might look Mondrianesque if one has been affected by Piet Mondrian’s abstract figurations of New York City. This concept has radical implications for models of perception and cognition, as well as for theories of aesthetics and representation. But it has rarely been stated systematically by art historians, though it is implied throughout their work — some of the best accounts come from art critics, such as Daniel-Henry Kahnweiler (who promoted Cubism), and writers such as Marcel Proust.
“If pictures change the way we naturally perceive the world around us, then many educational and other implications arise. As individuals, we benefit from being explicit about the visual assumptions we’re always making, often unaware of their origins in pictures we’ve seen. Collectively, when we try to recognize other people’s points of view — as a matter of social and moral responsibility — we’re possibly partly recognizing their pictorial experiences. Therefore, understanding their pictures is a crucial way of understanding their identities.”
— Whitney Davis
The Approach
The project Depictured Worlds: The Perceptual Power of Pictures will study “depicturation” from several points of view. The Depictured Worlds research team will identify likely art-historical cases of depicturation in the history of modern Western arts in their global dissemination, and build additional examples from indigenous, ancient, and/or non-Western arts. By conducting interviews with experts, the researchers will obtain perspectives from human evolution and prehistory, perceptual and cognitive psychology, and sociological and anthropological studies of visual culture. They also will investigate topics including the global dissemination of picture making in prehistory, the historical consequences of schematic and naturalistic styles of depiction, and the variety of social expectations for the perceptual power of pictures.
The project aims to provide a critical history of the concept of depicturation, to assess its usefulness in and implications for disciplines of visual studies in the widest sense, and to explore theoretical models and empirical investigations that might clarify it analytically and test it experimentally. Considering that the perceptual experiences of long-ago populations are not directly accessible, the researchers will explore algorithmic and/or computational approaches that might model complex processes of depicturation in a population.
Depictured Worlds is being led by Whitney Davis at the University of California, Berkeley (US).
Feature image: Brush-and-ink painting of the Derwentwater, England, by Chiang Yee, c. 1925. The art historian E. H. Gombrich used Chiang Yee’s pictures to argue that his perception of landmarks in Britain was shaped by his Chinese training. Is this reasonable?

Details
The Question
Celebrating victory — often depicted in a graphic and crude manner — is one of the most recurrent motifs in amateur soldier photographs of World War II, the largest global conflict to date and the first that was widely documented. Taking trophies from the battlefield is a universal and timeless human practice; as object and medium, however, photographs add an entirely new dimension to this phenomenon. Soldiers created and circulated photographic displays of triumph, transforming the experience of armed conflicts into a shared, intimate and eminently visual event.
The collaborative interdisciplinary research project Trophy Photographs: Performative Transgressions of Ordinary Soldiers in World War II considers “trophy photographs” taken by Axis and Allied soldiers in a transnational perspective as a distinct photographic genre and social practice that shaped not only the servicemen, but also their larger societies. Comprising 15 scholars, collectors and artists with diverse backgrounds, interests and specializations, the research team argues that trophy photographs hold historical, sociological and political meaning and reveal new aspects of essential social and cultural practices of warfare. Their inherently performative quality and evocative power transform trophy photographs into a weapon in their own right, with far-reaching consequences even today.
The Approach
The team’s collective effort to understand the history and meaning of trophy photographs in World War II requires reconsideration of the complex relationships between artifacts and actors; photographers and spectators; and past, present and future societies. As products of the interaction between their content (diegesis, or the space of the image) and their context (extradiegetic circumstances, or external and historical factors that can and do change), photographs not intended as trophies can become them.
Meanwhile, those photographs created to commemorate triumph might unintentionally become documents of atrocities. It is therefore necessary to explore the legacies of trophy photographs, as they construct alternative realities that soldiers and their communities later perceive as truth. Capable of keeping the past alive, trophy photographs often trigger contradictory memories and narratives, providing a sense of pride while also transmitting trauma across generations.
By examining World War II-era photographs and historical visual practices, the Trophy Photographs researchers are developing hermeneutical tools to dissect trophy photographs of past, current and future conflicts. The primary goal of this collaborative research is the creation of a photo textbook written for a broad audience that explores and reflects on the interplay between scholarly inquiry and the ethics of showing. This transdisciplinary approach aims to better comprehend present-day amateur war photographs circulating on social media and to generally deal with warfare in a more critical and conscious manner.
The Trophy Photographs project is being led by Elissa Mailänder at Sciences Po Centre for History in Paris, France, and the Centre Marc Bloch in Berlin, Germany, and by Tom Streuber at Tom, Dick & Harry GmbH in Berlin, Germany, and the Centre Marc Bloch. The research team includes Petra Bopp, Martin Dammann, Margaret Hillenbrand, Marianne Ingleby, Iain Johnston, Daniel H. Magilow, Regina Mühlhäuser, Malu Mühmer, Ulrich Prehn, Mary Louise Roberts, Sven Saaler, Yuki Tanaka, Jan Wenzel and Jialin Christina Wu.
Research is the vital expression of humankind’s most important qualities: curiosity and imagination.
Explorers, inventors, pioneers—dedicated researchers on the frontiers of science and the humanities.
Insight, when it comes, changes everything.