

NOMIS Awardee Adriano Aguzzi and colleague Elena De Cecco published an article in Science on Oct. 2, 2020, exploring the evolution of prions.

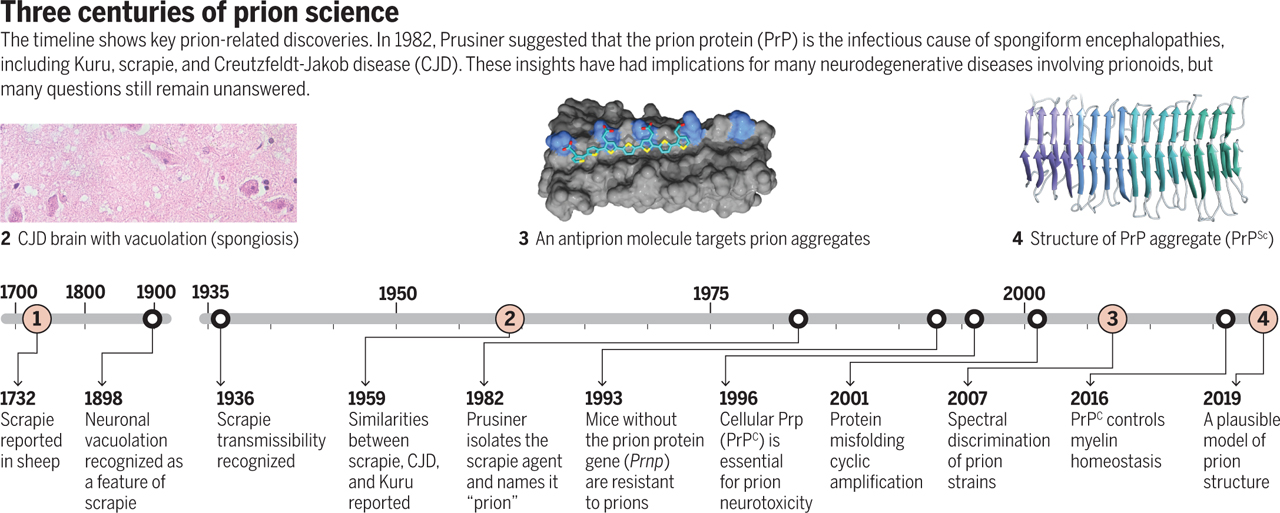
Paradigm shifts are drivers of scientific progress, yet the shifters of the paradigms often experience scorn rather than immediate applause. That was the fate of Stanley Prusiner’s 1982 paper claiming—to the initial amusement of his colleagues—that scrapie, a degenerative disease that affects the central nervous system of sheep, is caused by “proteinaceous infectious particles,” which he called prions (1). Prusiner’s intuition, which earned him the 1997 Nobel Prize, is influencing our approach to an ever-expanding variety of seemingly unrelated diseases and physiological processes, and its implications reverberate to the present day.
The two decades preceding Prusiner’s paper had witnessed the immense success of molecular biology, including the cracking of the genetic code; the elucidation of DNA replication, transcription, and translation; and the cloning of genes. These discoveries prompted Francis Crick to conceptualize the “central dogma”: Information flows unidirectionally from DNA to proteins. But although religious dogmas may be eternal, the shelf life of scientific dogmas is inevitably limited. Prusiner postulated that prions carry on their replicative cycle without the participation of nucleic acids. This hypothesis, reminiscent of John Griffith’s 1967 suggestion of the existence of self-replicating proteins (2), had the potential to explain the prodigious resistance of the scrapie agent to DNA-damaging radiation.
Daniel Carleton Gajdusek, who won a Nobel Prize for showing that Kuru was a human disease transmitted by cannibalism in Papua New Guinea, proposed in 1959 that the neurodegenerative disorders Kuru, scrapie, and Creutzfeldt-Jakob disease (CJD) are caused by “slow viruses.” Indeed, prions behave similarly to neurotropic viruses in many surprising ways, including the colonization of extraneural organs followed by neuroinvasion of the brain through peripheral nerves (3). Yet, Prusiner purified the agent and found it to be smaller than a virus: No informational nucleic acid would fit into it. Over time, the group of prion diseases grew to include other human (fatal familial insomnia) and animal (bovine spongiform encephalopathy, also called mad cow disease, and chronic wasting disease) disorders, but no causative virus has been identified and their prion etiology is now well accepted.
GRAPHIC: MELISSA THOMAS BAUM/SCIENCE; (IMAGES, LEFT TO RIGHT) K. FRONTZEK; U. S. HERMANN ET AL., SCI. TRANSL. MED. 7, 299RA123 (2015); G. SPAGNOLLI, ADAPTED FROM (12)
But prions did not contradict Crick’s central dogma after all. Charles Weissmann, refusing to believe that a protein could exist without its respective gene, discovered in hamsters the gene encoding the cellular prion protein (PrPC), whose misfolding yields tightly packed aggregates called PrPSc. It is generally believed that prion replication occurs when coalesced PrPSc is broken down into smaller species. Those species then accrue further PrPSc, in a process akin to the growth of crystals, and eventually break again, perpetuating their replicative cycle. Infectious prion seeds then move to neighboring cells and wreak havoc in the central nervous system by inducing vacuolation (“spongiosis”) within neurons (see the figure).
Does this mean that PrPSc is the prion? Weissmann’s discovery in 1993 that prion protein (Prnp)–ablated mice are resistant to scrapie (4) was designed to disprove the protein-only hypothesis but failed to do so. However, Prnp deletion in mice also fell short of proving the prion hypothesis. If PrPC were the receptor of an imaginary “scrapie virus,” its ablation may also render mice resistant to scrapie. More direct evidence for Prusiner’s ideas emerged in 2001 from Claudio Soto’s landmark experiment: Repeated cycles of PrPSc fragmentation, when followed by addition of PrPCand aggregate regeneration, can multiply prions ad libitum (5). These findings strengthen the hypothesis that the transfer of structural information can occur horizontally between proteins.
More recently, the prion concept has been applied, sometimes overenthusiastically, to virtually all diseases characterized by progressive deposition of aggregated proteins in the central nervous system, whether infectious or not—and even to physiological processes such as memory formation (6). α-Synuclein aggregates can self-propagate in the brains of Parkinson’s disease patients (7), in cultured cells, and in mice (8). This implies that a-synuclein is a de facto prion and that its handling demands high biosafety standards. Similar arguments were made for tau and amyloid-β (Aβ) aggregates, the major hallmarks of Alzheimer’s disease (9). However, prions caused many epidemics, whereas infectiousness has not been conclusively demonstrated for other protein aggregates—and specifically not through oral transmission. Protein aggregates that were not shown to be serially transmissible across multiple generations of hosts are better regarded as “prionoids,” even if they share molecular mechanisms of amplification with bona fide prions in vitro.
As predicted by Prusiner in the closing lines of his paper, the “prion revolution” boosted research in the field of neurodegeneration by providing an intellectual framework that might explain many aspects of Alzheimer’s disease, Parkinson’s disease, and many other neurodegenerative diseases featuring protein aggregates. Although cellular PrPC is now known to be crucial for the maintenance of peripheral myelin (10), our understanding of prions has essentially stagnated for more than a decade and may now be lagging behind that of prionoids. What is really known about prions, after almost 40 years since Prusiner’s discovery?
Continue reading this Science publication
